By Gene Vantage – 20 September 2024
Polymerase Chain Reaction (PCR) is a powerful and widely used technique in molecular biology for amplifying specific segments of DNA. Developed by Kary Mullis in the 1980s, PCR has revolutionized genetics, diagnostics, and various fields of biological research. The process involves three basic steps—denaturation, annealing, and extension—repeated over multiple cycles to exponentially amplify the target DNA sequence. Successful PCR relies on carefully balancing several critical components, including template DNA, primers, dNTPs (deoxynucleotide triphosphates), DNA polymerase, buffer, and MgCl₂. This guide provides a concise overview of how to optimize these variables for achieving specific, efficient, and high-yield amplification.
1. Template DNA: Finding the Right Balance
The quality and concentration of your template DNA can make or break your PCR reaction:
Concentration Matters: It is recommended to start with 1–10 ng for plasmid DNA, 50–500 ng for genomic DNA, and 1–100 ng for cDNA per 50 μL reaction. If you observe weak amplification, increase the DNA concentration. If you see non-specific bands, try lowering it by doing a dilution series. A good dilution series would be to dilute your DNA in a 1:10 and 1:20 ratio and load the same volume as you would a normal PCR to try and dilute out inhibitors.
Quality is Key: Ensure your DNA is free from inhibitors and contaminants like proteins, salts, or organic solvents. High-quality, intact DNA results in more reliable amplification and low-quality DNA can inhibit the reaction. Consider additional purification steps if necessary.
Adjust for Complexity: For complex templates like genomic DNA, you might need higher concentrations, longer extension times, or specialized enzymes that can handle complex DNA more efficiently.

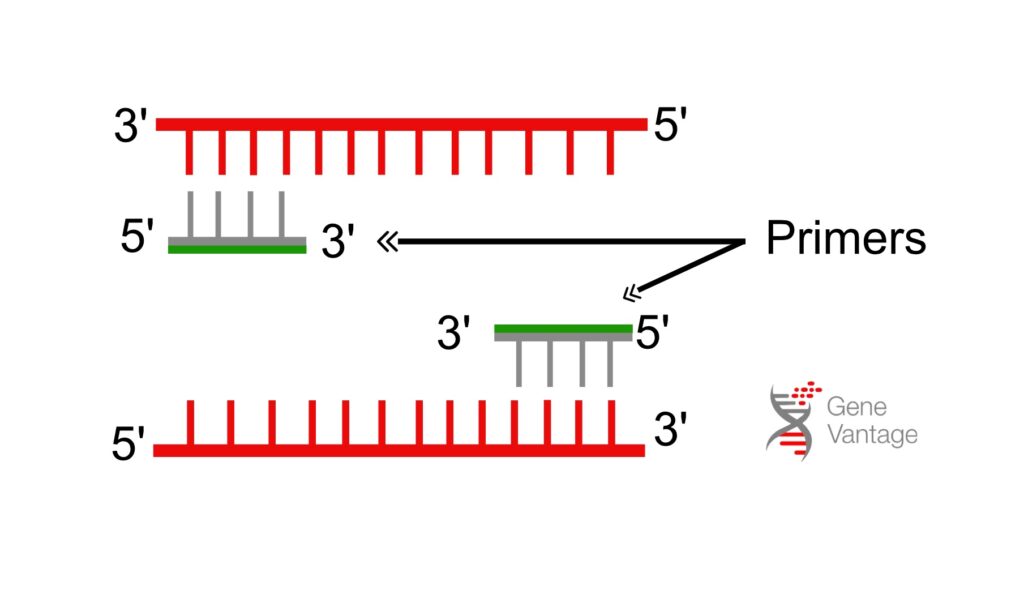
2. Primer Design and Concentration: The Foundation of Specificity
Primers play a pivotal role in determining the specificity and efficiency of PCR. Both the design and concentration of primers must be mindfully optimized:
Concentration Optimization: Begin with 0.2 μM for each primer as a starting point. If primer-dimers or non-specific products form, reduce the concentration in 0.05 μM steps. If the amplification yield is too low, increase it up to 0.5 μM.
Design Considerations: Primers should usually be between 18–25 bases long with a GC content of 40-60%. Avoid sequences that form secondary structures (like hairpins) or have repetitive regions that could cause off-target amplification.
Melting Temperature (Tm): Choose primers with similar Tm values (within 2–3°C) to ensure efficient binding. Conduct a gradient PCR (if possible) to identify the optimal annealing temperature, typically 3–5°C below the primers’ Tm.
3. Balancing dNTP and MgCl₂ Concentrations for Effective Amplification
Deoxynucleotide triphosphates (dNTPs) are the building blocks of DNA synthesis. MgCl₂ Acts as a cofactor that enhances the activity of DNA polymerases. Their concentration can influence the fidelity and efficiency of PCR:
dNTPs: Start with 200 μM of each dNTP. If you observe smearing or non-specific bands, ensure that dNTPs are fresh and of high purity. For longer templates or high-fidelity enzymes, you may increase the concentration to 250–300 μM.
MgCl₂: Magnesium ions are essential for polymerase activity and DNA-DNA duplex stability. Begin with 1.5 mM MgCl₂ and adjust in 0.25–0.5 mM increments. Higher MgCl₂ concentrations can increase yield but may reduce specificity. Remember that higher dNTP or primer concentrations may chelate Mg²⁺, requiring a corresponding increase in MgCl₂.
4. Choosing the Right DNA Polymerase
The choice of DNA polymerase depends on the application and template complexity:
Standard Taq Polymerase: Suitable for routine PCR where fidelity is not the primary concern.
High-Fidelity Enzymes (e.g., Q5, Phusion, Pfu): These are ideal for applications that require high accuracy, such as cloning or sequencing, due to their proofreading ability.
Hot-Start Polymerases: These enzymes remain inactive at room temperature and only activate at elevated temperatures, preventing non-specific amplification and primer-dimer formations.
Concentration Adjustments: Standard concentration is 1–2.5 U per 50 μL reaction. Too much enzyme can lead to non-specific bands, while too little may result in weak or no amplification. Adjust enzyme concentration based on observed results.
5. Buffer Composition and Additives: Fine-Tuning the Reaction Environment
The PCR buffer provides a stable pH and the ionic conditions required for optimal enzyme activity:
Use the Recommended Buffer: Each polymerase comes with an optimized buffer. Use it as provided, especially for high-fidelity enzymes where buffers are designed to maximize accuracy.
Additives to Enhance Performance:
- DMSO (Dimethyl Sulfoxide): Effective for GC-rich templates; start with 5% and adjust up to 10%.
- Betaine: Helps in reducing secondary structures in difficult templates; start at 1 M and adjust up to 2.5 M.
- BSA (Bovine Serum Albumin): Stabilizes enzymes, particularly in challenging samples; use at 0.1 mg/mL.

6. Fine-Tuning Cycling Conditions: Optimizing PCR Steps
Each stage of the PCR cycle can be optimized to improve specificity and yield:
Denaturation Step: Use 94–98°C for 20–30 seconds to melt the DNA strands. Avoid extended high-temperature exposure to prevent DNA degradation.
Annealing Temperature: Perform a gradient PCR (e.g., 55–70°C) to determine the optimal annealing temperature, typically 3–5°C below the primer Tm. Adjusting the annealing temperature by as little as 1-2°C can significantly affect specificity and yield.
Extension Step: For Taq polymerase, use 72°C with 30 seconds per kb of DNA. For high-fidelity enzymes, follow the manufacturer’s recommended temperatures and times, as these can vary.

7. Adjusting Reaction Volume and Instrument Settings
The reaction volume and thermal cycler settings also play a role in cost and optimization of PCR reactions:
Reaction Volume: The standard is 25–50 μL. Smaller volumes (10–20 μL) can save reagents but may require re-optimization due to changes in reaction dynamics.
Thermal Cycler Settings:
- Ramp Rates: Slower ramp rates (2–3°C/sec) can improve specificity by allowing more precise primer binding, although they increase run time.
- Lid Temperature: Set the lid to 105°C to prevent condensation and ensure consistent reaction conditions.
8. Use Controls to Validate Your PCR Setup
Always include controls to confirm the reliability of your PCR results:
- Negative Control: Include a no-template control to check for contamination in reagents or equipment.
- Positive Control: Use a known template to ensure all reaction components are working properly and that the PCR setup is correct.
Conclusion: A Systematic Approach to PCR Optimization
Optimizing PCR is a step-by-step process that involves adjusting one variable at a time, starting with standard conditions. Fine-tune each component to achieve the highest specificity and yield for your application. By carefully considering each variable and making deliberate adjustments, you can consistently achieve high-quality amplifications tailored to your research needs.
For more in-depth tips or troubleshooting guidance, feel free to reach out to the Gene Vantage technical support team. Happy amplifying!